









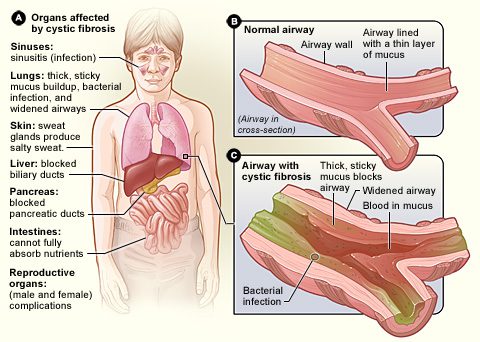
Cystická fibróza (cystická fibróza)
La cystická fibróza, Je zde genetické onemocnění nejčastější. Hlavní projevy se týkají dýchacího a trávicího traktu, ale mohou být postiženy téměř všechny orgány. Symptomy se často objevují v raném dětství a liší se v závažnosti od člověka k člověku. Toto onemocnění způsobuje a zahušťování hlen vylučovaný sliznicemi dutin, průdušek, střeva, slinivky břišní, jater a reprodukčního systému (viz schéma).
Projekt plíce jsou často nejvážněji postiženy. The husté, viskózní sekrety ucpávají průdušky, což ztěžuje dýchání. Kromě toho hlen, který se hromadí v plicích, přispívá k růstu choroboplodných zárodků. Lidé s cystickou fibrózou jsou proto více ohroženi častými a potenciálně závažnými respiračními infekcemi.
La cystická fibróza se také dotýká zažívací ústrojí. Hlen má tendenci blokovat tenké kanálky slinivky břišní, čímž brání trávicím enzymům produkovaným slinivkou ve vstupu do střeva a vykonávání jejich činnosti. Vzhledem k tomu, že potrava je trávena jen částečně, zejména tuky a některé vitamíny, dochází k jejich značným nedostatkům. Mohou vést k a zpomalení růstu.
Toto onemocnění má také závažné dopady na játra a reprodukční orgány, což často vede k subfertilitě u žen a neplodnost u postižených mužů.
Díky dřívější diagnóza a lepší péči,životní očekávání a kvalita života postižených se v posledních desetiletích neustále zlepšuje, zejména poté, co se začínají objevovat nové terapie zaměřené na genetickou anomálii, které ve střednědobém horizontu změní léčbu pacientů. .
Prevalence
La cystická fibróza je genetické onemocnění nejběžnější ve Francii s téměř 6000 postiženými lidmi1.. Touto nemocí trpí jeden ze 4 novorozenců. Mnohem vzácnější je mezi černochy (000 v 1) a orientálci (13 v 000). Postihuje muže i ženy. Nejvíce je postiženo obyvatelstvo západní Francie.
La cystická fibróza je genetické onemocnění nejběžnější závažné onemocnění v Kanadě. Postiženo je jedno ze 3 novorozenců1. Cystická fibróza je o něco častější u Quebec než ve zbytku Kanady: postiženi jsou 3 Kanaďané, včetně 500 Quebečanů.
Příčiny
La cystická fibróza byl poprvé popsán v roce 1936 Dr Guido Fanconi, švýcarský pediatr. Odpovědný gen, pojmenovaný CFTR (pro „regulátor transmembránové vodivosti cystické fibrózy“), nebyl identifikován až do roku 1989 kanadskými výzkumníky. U nemocných lidí tohle gen is abnormální (říkáme, že je převeden). Je zodpovědný za syntézu chlórového kanálu umožňujícího regulovat hydrataci hlenu. V případě abnormality v genu CFTR, sliz produkt je příliš hustý a nestéká normálně. Byla identifikována více než 1 různá mutace v genu CFTR zapojená do cystické fibrózy2, 3,4. Jsou rozděleny do 6 tříd podle různého typu dysfunkce2Z těchto mnoha mutací je nejčastější mutace Delta F508, která se vyskytuje u 81 % postižených lidí ve Francii.
Cystická fibróza není nakažlivé onemocnění. Lidé, kteří vlastní patogenní mutace genu CFTR vyvine onemocnění dříve nebo později, ale v různé míře závažnosti.
Diagnostický
Obvykle je cystická fibróza diagnostikována již v prvním roce života, protože respirační příznaky objevují velmi brzy. V 90 % případů je onemocnění zjištěno před dosažením věku 10 let.
K potvrzení diagnózy lékař provede a potní test (nebo potní test). Ve skutečnosti je pot lidí s cystickou fibrózou mnohem více koncentrovaný v soli (2 až 5krát více než normálně). The genetické testy umožňují přesnou identifikaci abnormalit v genu CFTR. Jsou nezbytné pro zvážení cílené terapie.
Ve Francii je cystická fibróza systematicky vyšetřována u všech novorozenců od roku 20025. Bylo prokázáno, že včasný screening zlepšuje kvalitu života a očekávanou délku života postižených dětí. Novorozenci se odebírají ve 3 dnech života po souhlasu rodičů, před propuštěním. mateřství. Test nedává definitivní diagnózu, ale ta bude potvrzena nebo vyvrácena specifickými doplňkovými vyšetřeními (potní test, genetická studie).
V Quebecu neexistuje systematický screening tohoto onemocnění. Kanadská nadace pro cystickou fibrózu podporovaná několika lékaři však již několik let volá po zavedení novorozeneckého screeningu. Ukázalo se, že včasná detekce zlepšuje kvalitu života a délku života postižených dětí.
Naděje dožití
V sadě 1960sživotní očekávání dětí s cystickou fibrózou nepřesáhlo 5 let. V současné době je podle posledních statistik střední věk přežití 47 let1. respirační infekce zůstávají nejčastější příčinou úmrtí.
Časté komplikace
Cystická fibróza je onemocnění, které postupně poškozuje plíce, slinivku a játra. a lékařské monitorování Pomáhá však snižovat závažnost a četnost komplikací.
Projekt respirační komplikace jsou nejčastější, včetně dilatace průdušek způsobující bronchitidu, zápal plic s opakováním. Jsou období zhoršení respiračních příznaků, kdy jsou pacienti velmi „přetížení“, více se zadýchávají, hubnou, často kvůli infekci. Poškození dýchacích cest může být život ohrožující.
Pokud jde o zažívací ústrojí, obstrukce žlučových cest, které umožňují žluči proudit do trávicího traktu, může vést k cirhóze jater. Obstrukce a progresivní skleróza slinivka břišní, může způsobit malabsorpci živin a rozvoj diabetu. Tyto poruchy často vedou k nutriční nedostatky těžký a chronický průjem. Obecně lze nedostatky napravit speciální dietou. Může se naopak objevit i výrazná zácpa, nebo dokonce střevní neprůchodnost.
Obvykle se puberta objevuje později u chlapců a dívek s cystickou fibrózou. Konečně, plodnost je snížena, zejména u mužů, kteří jsou téměř všichni (95 %) sterilní v důsledku obstrukce chámovodu. Tyto kanálky přenášejí spermie z varlat do semenných váčků. U žen zvýšená viskozita vaginálního hlenu zpomaluje pohyb spermií. Onemocnění může ovlivnit i pravidelnost a frekvenci ovulace. Plodnost klesá, ale těhotenství je stále docela možné.